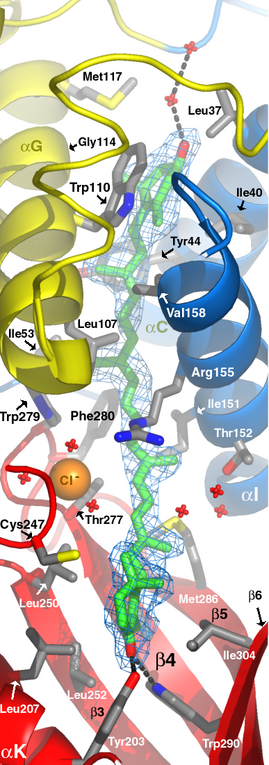
这次附了两个图: 1)snap3 shows the N-C-H...O hbond longer than 4A, which is claimed be to critical in that structure. 2)snap4 shows that mutation I was talking about. See that hydrogen bond (3. 13A) is between two sheets. If we mutate that to non-polar aa, can you give any speculations? 3)SNAP5 是我自己用vmd represent的electrostatic potential surf 和peptide binding的图(非常感谢前面回帖的所有人,我总算蜗牛般地学了点vmd). snap6是文献里面的同样的展示内容. 困扰我的是我做的里面那个binding site像个闭合的孔,而文献里面四个半开放的 channel. 我的surf不是因为计算的electrostaic potential的问题,因为如图SNAP7,我即使只做 surf,而不要potential,那个binding site 还是闭合的.能不能指点一下问题所在? 4)追问一下:homology modelling,在 modeller,jmol,spdv,vmd这几个里面,哪个更被专业的人认可(活着jmol和vmd根本没有这个功 能?)? 非常非常感谢
【在 s********9 的大作中提到】 : 这次附了两个图: : 1)snap3 shows the N-C-H...O hbond longer than 4A, which is claimed be to : critical in that structure. : 2)snap4 shows that mutation I was talking about. See that hydrogen bond : (3. : 13A) is between two sheets. If we mutate that to non-polar aa, can you : give : any speculations? : 3)SNAP5 是我自己用vmd represent的electrostatic potential surf 和peptide : binding的图(非常感谢前面回帖的所有人,我总算蜗牛般地学了点vmd).
s*9
33 楼
【在 s********9 的大作中提到】 : 这次附了两个图: : 1)snap3 shows the N-C-H...O hbond longer than 4A, which is claimed be to : critical in that structure. : 2)snap4 shows that mutation I was talking about. See that hydrogen bond : (3. : 13A) is between two sheets. If we mutate that to non-polar aa, can you : give : any speculations? : 3)SNAP5 是我自己用vmd represent的electrostatic potential surf 和peptide : binding的图(非常感谢前面回帖的所有人,我总算蜗牛般地学了点vmd).

{kind=link}