海归学者发起的公益学术平台
分享信息,整合资源
交流学术,偶尔风月

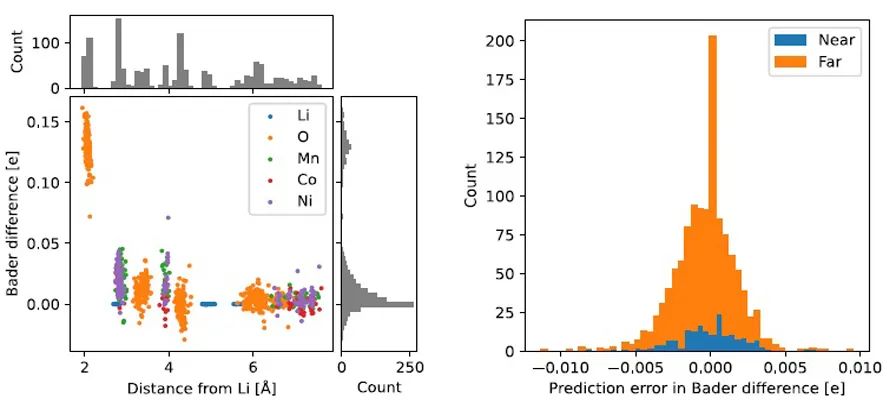
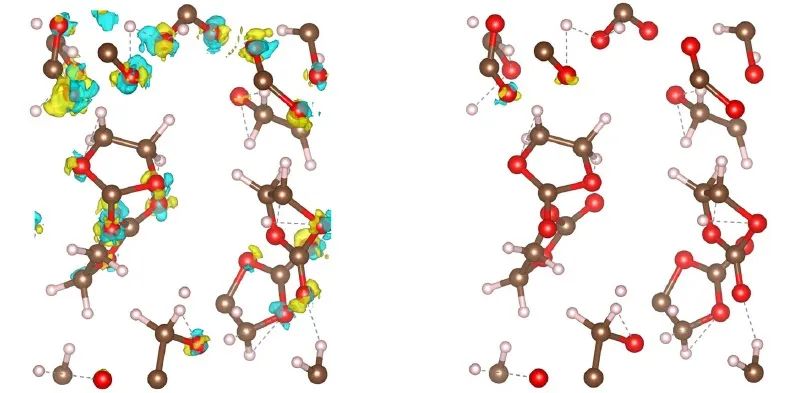
计算和实验对当代材料研究同样重要,如基于量子力学的原子尺度模拟经常用于新型功能材料与分子的计算搜索,其中最典型的方法就是密度泛函理论DFT。电子密度ρ(r)是DFT计算中至关重要的基本变量,因为ρ(r)可以决定体系的基态性质。然而,DFT框架往往存在计算尺寸问题,且难以探索巨大的相空间。因此,人们开始发展基于量子力学的机器学习势。早期的机器学习势往往是用于体系总能的预测模拟,无法很好地给出电子结构或者ρ(r)依赖的信息。近年来有部分机器学习工作是针对ρ(r)的预测,以核回归方法为主,但是该类方法的计算复杂度会会随着训练数量呈立方关系增长。因此,人们开始利用深度神经网络来进行大规模数据集的训练及后续的预测,如DeepDFT,它可以进行逐点的密度预测。然而,这种方法无法通过传递来解析角度信息。
来自丹麦科技大学能源转换与存储学院的Peter Bjørn Jørgensen和Arghya Bhowmik等人,基于原有的神经网络DeepDFT模型,通过引入等变信息传输,构建了等变图神经网络,即等变DeepDFT。该方法可以有效地传递角度信息,并且完全是数据驱动的,不需要任何的人为操控,能有效、准确地预测出体系的电子密度ρ(r)。该模型在QM9分子、金属氧化物锂离子电池阴极材料,以及液态碳酸乙酯电解质分子动力学等数据库进行了测试,结果表明了等变图神经网络方法具有很好的普适性。该工作指出,与氧化还原反应和电荷转移特性有关的高通量筛选和模拟,会在不久的将来通过这种方法得到解决。由于该代码可供学术界使用,作者期望看到该方法在材料和分子模拟中得到广泛应用,以及在其它模型中进行扩展。
该文近期发表于npj Computational Materials 8:183(2022),英文标题与摘要如下,点击左下角“阅读原文”可以自由获取论文PDF。
Equivariant graph neural networks for fast electron density estimation of molecules, liquids, and solids
Peter Bjørn Jørgensen & Arghya Bhowmik
Electron density ρ(r) is the fundamental variable in the calculation of ground state energy with density functional theory (DFT). Beyond total energy, features and changes in ρ(r) distributions are often used to capture critical physicochemical phenomena in functional materials. We present a machine learning framework for the prediction of ρ(r). The model is based on equivariant graph neural networks and the electron density is predicted at special query point vertices that are part of the message-passing graph, but only receive messages. The model is tested across multiple datasets of molecules (QM9), liquid ethylene carbonate electrolyte (EC) and LixNiyMnzCo(1-y-z)O2 lithium ion battery cathodes (NMC). For QM9 molecules, the accuracy of the proposed model exceeds typical variability in ρ(r) obtained from DFT done with different exchange-correlation functionals. The accuracy on all three datasets is beyond state of the art and the computation time is orders of magnitude faster than DFT.