海归学者发起的公益学术平台
分享信息,整合资源
交流学术,偶尔风月

分子和原子在固态材料表面的化学吸附是化学、物理和材料科学中的一个中心概念。识别表面和吸附物的性质,从而能控制化学吸附的强度,对于理解表面科学中的化学过程至关重要。过渡金属元素、过渡金属合金和金属间化合物是催化领域中重要的一类材料。研究人员对这些系统已经进行了大量的研究,并提出了一些简单的模型来描述这些表面上的相互作用。最常见的是由Hammer & Nørskov 提出的d带中心模型,它有效地将在衬底和吸附物相互作用之前的电子结构特征与化学吸附强度关联起来。d带中心模型利用d带相对于费米能级的位置,解释了纯过渡金属和某些合金的吸附趋势。然而,对很多合金和金属间化合物,d带中心模型不能描述由合金引起的电子结构不对称性和变形。此外,目前预测准确化学吸附能量一般是基于数据驱动的方法,这表明一旦将材料空间扩展到简单的单金属或双金属系统外,就会遇到与组合复杂性相关的巨大的挑战。
来自美国SLAC国家国家加速器实验室SUNCAT界面科学与催化中心的Frank Abild-Pedersen教授等人,提出了一种化学吸附模型,考虑了吸附物引起的表面位点扰动。该模型成功地解释了调控活性位点(活性位点周围化学环境发生变化)时所观察到的化学吸附强度的变化。此外,该模型准确地描述了O、N、CH和Li等吸附物,在由Au、Ag、Cu和Pt与3d、 4d和5d金属组成的双金属/三金属合金表面的吸附能,平均绝对误差达到0.13 eV。对于给定的吸附衬底,作者仅用了四个参数对模型进行了参数化,即d带的一阶和二阶矩以及相邻原子的d带填充。作者根据表面特征有效地预测化学吸附能,并可以利用少量的DFT训练数据就能优化特定吸附物和吸附位点的模型参数。该方法已推广到了整个过渡金属系列和多组分体系,可用于指导具有理想催化性能的复杂合金的工程化研究。
该文近期发表于npj Computational Materials 8:163(2022),英文标题与摘要如下,点击左下角“阅读原文”可以自由获取论文PDF。
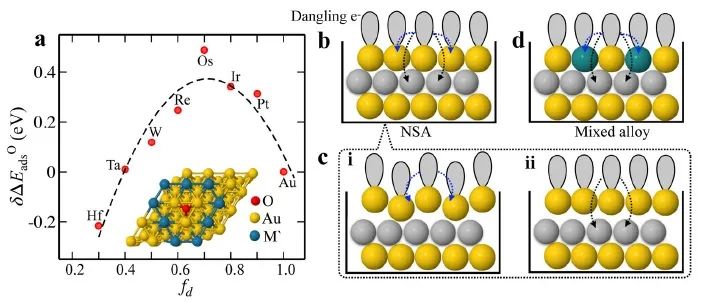
Electronic structure factors and the importance of adsorbate effects in chemisorption on surface alloys
Shikha Saini, Joakim Halldin Stenlid & Frank Abild-Pedersen
The chemisorption energy is an integral aspect of surface chemistry, central to numerous fields such as catalysis, corrosion, and nanotechnology. Electronic-structure-based methods such as the Newns-Anderson model are therefore of great importance in guiding the engineering of material surfaces with optimal properties. However, existing methods are inadequate for interpreting complex, multi-metallic systems. Herein, we introduce a physics-based chemisorption model for alloyed transition metal surfaces employing primarily metal d-band properties that accounts for perturbations in both the substrate and adsorbate electronic states upon interaction. Importantly, we show that adsorbate-induced changes in the adsorption site interact with its chemical environment leading to a second-order response in chemisorption energy with the d-filling of the neighboring atoms. We demonstrate the robustness of the model on a wide range of transition metal alloys with O, N, CH, and Li adsorbates yielding a mean absolute error of 0.13 eV versus density functional theory reference chemisorption energies.