
结直肠癌在全球范围内属于高发癌症。美国癌症协会估计美国2022年新发结直肠癌病例超过15万。根据国家癌症中心最新数据,我国结直肠癌发病率跃升至第二位,仅次于肺癌,每年新发的结直肠癌患者超过40万。结肠癌在结直肠癌中最为普遍,但目前我们还没有探明结肠癌的分子机制。 自噬是一种仅发生在真核细胞中的重要的代谢过程。它使得溶酶体降解异常和代谢失调的蛋白质和细胞器并且为细胞提供营养和能量。已有研究表明,自噬参与和细胞死亡密切相关的多种生理过程,并参与许多信号通路。足够的证据表明,自噬可以通过Bcl-2, EGFR或其他信号通路,促进或抑制肿瘤生长。好啦,铺垫的部分我们分别介绍了今天要复现的文章里研究的疾病和表型:结肠癌和自噬,下面就要进入正题了。今天带来的这篇文章,题目为“Study on the Expression Profile of Autophagy-Related Genes in Colon Adenocarcinoma “,是一篇2022年5月发表在Computational and Mathematical Methods in Medicine上的生信分析文章,全文基本都是可以使用仙桃工具进行分析的,没有加任何实验。虽然分数稍微低了一些,如果稍微完善一下其实还可以尝试投更高分数的期刊。话不多说,我们一起来看一下吧~~题目:Study on the Expression Profile of Autophagy-Related Genes in Colon Adenocarcinoma期刊:Computational and Mathematical Methods in Medicine

技术路线: 作者从the Human Autophagy Portal数据库(http://www.autophagy.lu/index HTML)下载了人类基因组全部232个自噬相关基因(ARGs),从TCGA数据库提取结肠癌中232个自噬相关基因(ARGs)的RNA表达谱和临床数据,包含398个肿瘤样本以及39个正常样本。GO富集分析的数据来源于Cistrome project (http://www.cistrome.org/)。KEGG富集分析的数据来源于KEGG project(http://www.kegg.jp/)。自噬相关基因(ARGs)的RNA表达谱数据经归一化后,使用R语言的limma包分析表达差异,阈值|logFC|>1,padj<0.05,得到的差异表达的ARGs和相关的临床数据用于后续分析。为了探求ARGs的肿瘤相关的潜在分子机制,采用R语言注释GO功能并进行KEGG富集分析。接着作者构建了预后模型:首先用Cox单因素回归分析了结肠癌和正常样本中38个差异表达的ARGs,结果显示24个ARGs和预后显著相关。接着进行Cox多因素回归, 8个ARGs为独立预后因子。之后K-M曲线评价高/低风险组的统计学差异并用ROC曲线评估模型的区分度。3.图表简介
Figure 1| ARGs在结肠癌和正常样本中的差异表达(Figure 1a| ARGs表达热图(Tumor vs Normal),Figure 1b| ARGs表达火山图(Tumor vs Normal),Figure 1c| ARGs表达分组比较图(Tumor vs Normal))
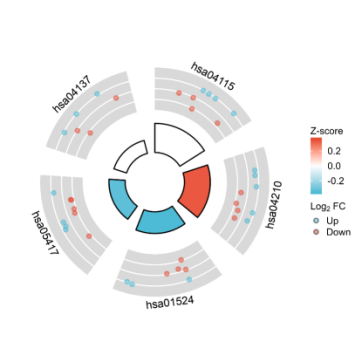
Figure 2| 差异表达ARGs的GO/KEGG富集分析(Figure 2a| GO富集分析气泡图,Figure 2b| GO联合logFC富集分析-气泡图,Figure 2c| KEGG联合logFC富集分析-圈图)
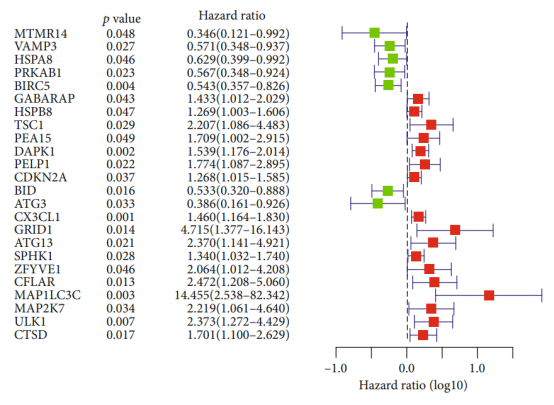
Figure 3| 差异表达ARGs的森林图
Figure 4| 预后模型的可视化-风险因子图(Figure 4a| ARGs在高/低风险组中的表达热图,Figure 4b| 高/低风险组中风险得分分布,Figure 4c| A高/低风险组中的生存时间,Figure 4d| ARGs表达分组比较图(高/低风险组))
Figure 5| 预后模型的评价(Figure 5a| 预后模型-列线图(基于Cox单因素回归结果),Figure 5b| 预后模型-列线图(基于Cox多因素回归结果),Figure 5c| 预后模型-K-M生存曲线,Figure 5d| 预后模型-ROC曲线)
Figure 6| 临床特征分组比较图(预后模型筛选出的显著影响预后的因子和临床特征的相关性)。
4.分析工具
5.复现流程
Figure 1| ARGs在结肠癌和正常样本中的差异表达Figure 1a| ARGs表达热图(Tumor vs Normal);Figure 1b| ARGs表达火山图(Tumor vs Normal);Figure 1c| ARGs表达分组比较图(Tumor vs Normal))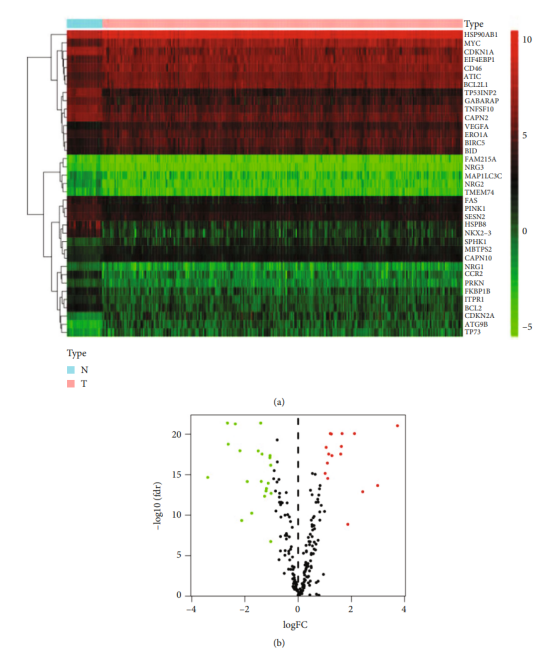
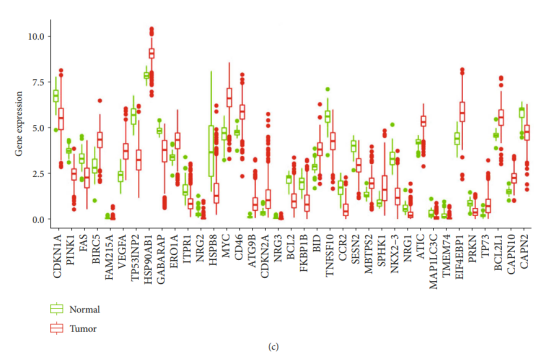
Figure 1b| ARGs表达火山图(Tumor vs Normal)复现:第1步:从Human Autophagy数据库(http://www.autophagy.lu/index HTML)下载自噬相关基因(ARGs)列表。登录Human Autophagy数据库(http://www.autophagy.lu/index HTML)→点击【Clustering】→复制从A到Z的全部ARGs列表→粘贴到新的Excel文件中→保存文件。
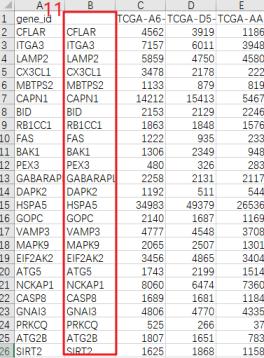
第2步:通过仙桃网盘下载TAGA-COAD RNA-seq COUNT数据下载的TAGA-COAD RNA-seq COUNT数据是txt格式文件,用Excel打开后如下所示:
PS:Count数据是原始测序得到的,count数就是比对到某个基因i上的总数目,是一个整数值。第3步: 从第2步下载的TCGA-COAD RNA-seq COUNT数据中提取ARGs表达谱。整理ARGs列表,只保留最后一列Gene Symbol数据→用Excel打开TCGA-COAD RNA-seq表达谱并另存为TCGA-COAD-ARGs→在第一列gene_id的右侧新建一列→用【VLOOKUP函数】精确匹配第1步得到的ARGs列表→数据筛选去掉【#NA】的项→选中整张工作表用【Ctrl+G】定位到当前区域,复制并粘贴到新的Excel文件→删除第2列→在当前工作表中选中第1列,用【条件格式】查找重复值,并删除整行都是空值的行→另存为CSV格式文件,得到了TCGA-COAD-ARGs表达谱数据。
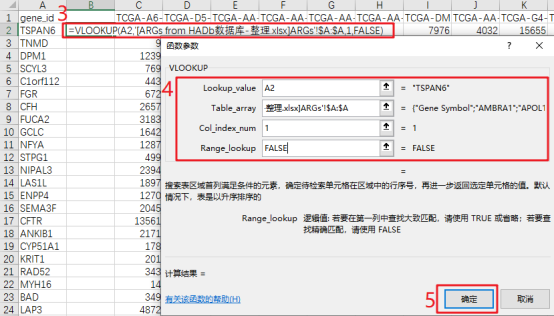
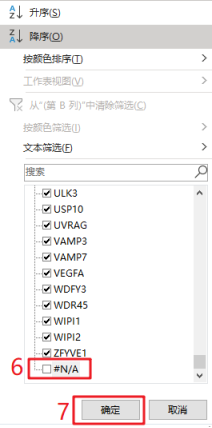
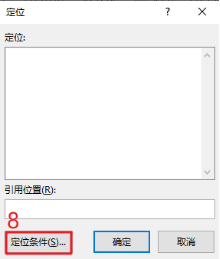
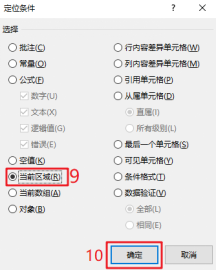
CSV格式文件只能保存当前工作表,如果需要建立多个工作表,选择Excel工作簿。根据样本名称区分Tumor/Normal组,例如TCGA-A6-5664-01A-21R-1839-07,第14个字符开始的2位是01,根据样本命名规则01~09是癌症样本,10-19是正常样本,20-29是癌旁样本。复制TCGA-COAD-ARGs表达谱第一列数据,转置粘贴到新建的Excel工作表→第2列中用MID函数提取样本名称的第4段→复制第2列并选择性粘贴数值到第3列,对第3列用IF函数筛选,标记Tumor/Normal组。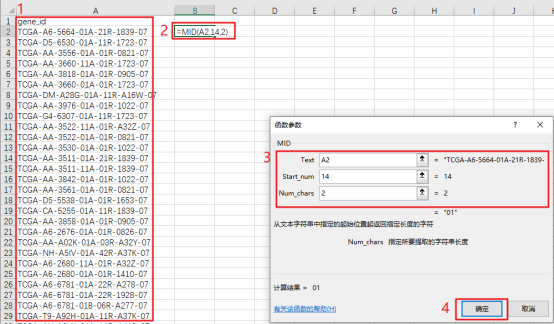
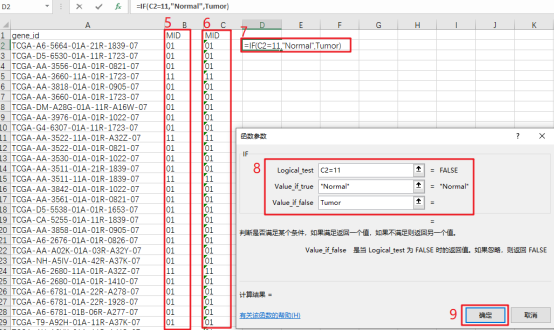
我们看到所有的样本都被标记为为Tumor,是因为第3列的单元格前面有绿色小三角,为文本格式。筛选出第3列为11的项,双击单元格,转为数据格式后,标记由Tumor变为Normal。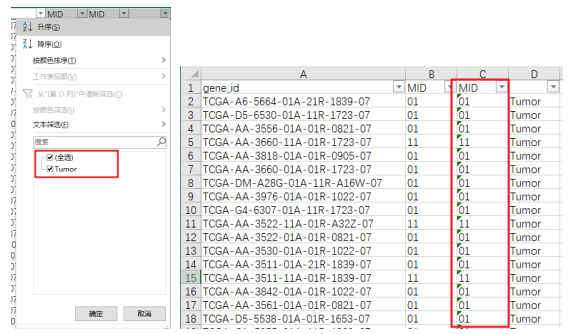
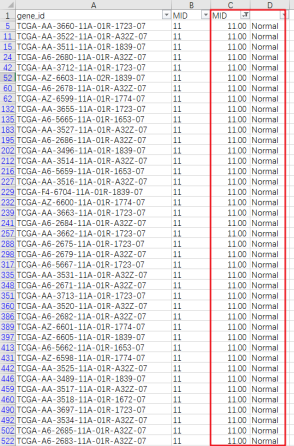
复制第1列和第4列并选择性粘贴数值到新的工作表中,输入第2列列名为group→删除第一个工作表,样本信息就准备好了。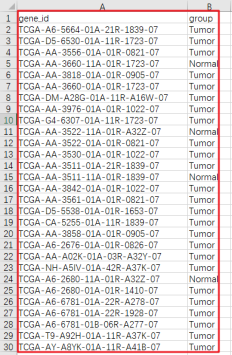
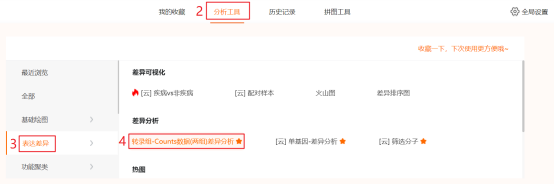
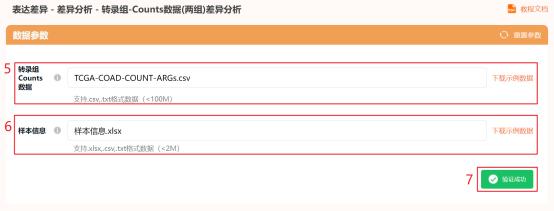
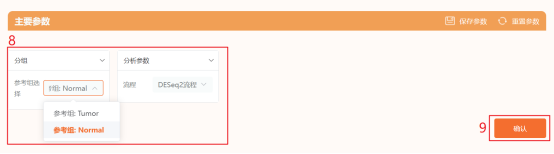
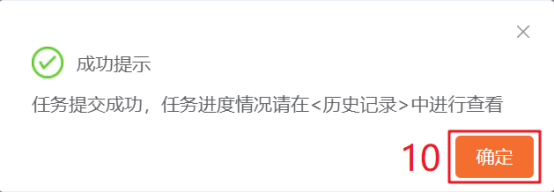
进入仙桃学术(https://www.xiantao.love/)(新版)→选择【生信工具】→点击上方工具选择栏【分析工具】→点击左侧导航栏【表达差异】→点击【转录组-Counts数据(两组)差异分析】→上传第3步和第4步准备好的表达谱和样本信息数据→点击【验证】→【主要参数】中【参考组】选择【Normal】,【分析流程】默认【DEseq2流程】→点击【确认】,出现任务提交成功提醒。完成分析可能需要几分钟时间,之后可以点击顶部工具栏的【历史记录】,查询和下载分析结果。

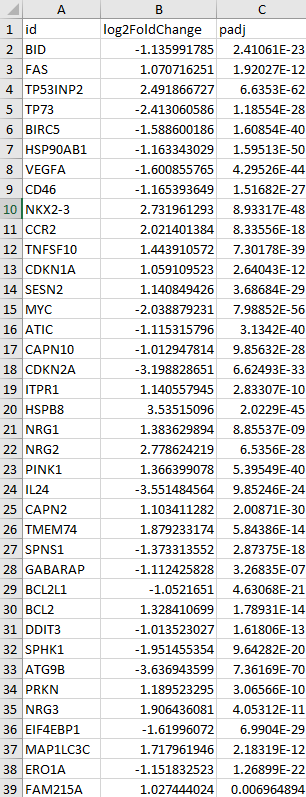
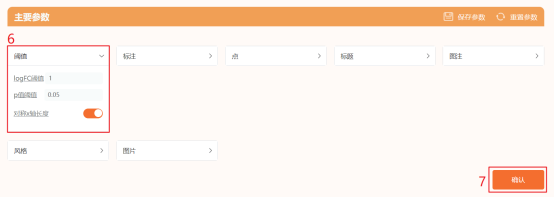
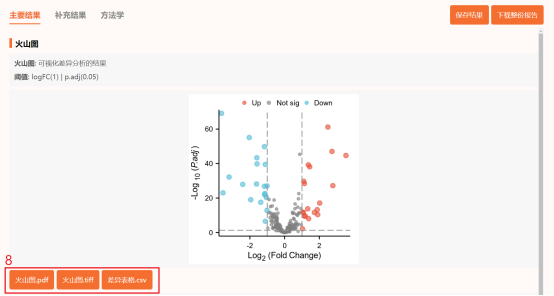
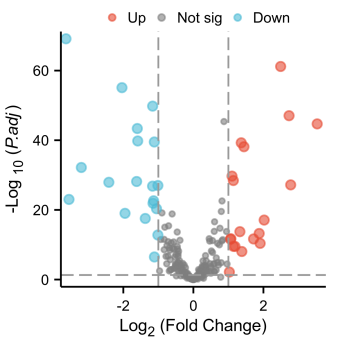
点击上方工具选择栏【分析工具】→点击左侧导航栏【表达差异】→点击【火山图】→上传第6步整理好的数据→点击【验证】→【主要参数】中【阈值】默认【logFC阈值】为1,【P值阈值】为0.05→默认其他主要参数→点击【确认】→下载【差异表格.csv】和火山图图片。这样就完成了Figure 1b的复现。
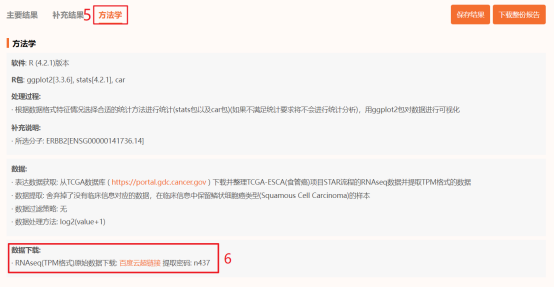
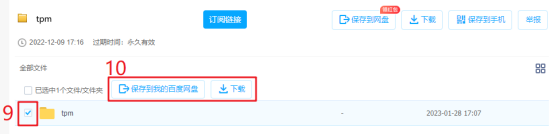
第1步:通过仙桃学术下载TPM格式的TCGA-COAD RNA-seq表达谱数据点击工具选择栏【分析工具】→点击左侧导航栏【表达差异】→点击右侧【 [云]疾病vs非疾病】→默认所有参数→点击【确认】→点击【方法学】→点击【百度云超链接】并复制提取码→在新窗口中粘贴提取码并点击【提取文件】→选择文件并点击【下载】。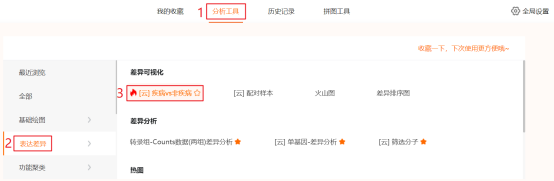

该文件包含了33种TCGA肿瘤的TPM格式的RNA-seq表达谱数据,整体下载时间会很长,建议先把文件保存到百度网盘,下载其中的TCGA-COAD RNA-seq表达谱数据。
下载的TPM格式的TCGA-COAD RNA-seq表达谱数据是txt格式。之后需要编辑时用Excel打开即可。
第2步,参考Figure 1b绘制的第3步, 从第1步下载的TPM格式的TCGA-COAD RNA-seq数据中提取【差异分析表格】中标记为Up和Down的差异表达ARGs的表达谱。PS:这一步【VLOOKUP】函数需要精确匹配的是差异表达的ARGs,而非全部ARGs。因此需要将筛选Up和Down的差异表达ARGs并复制到新的工作表。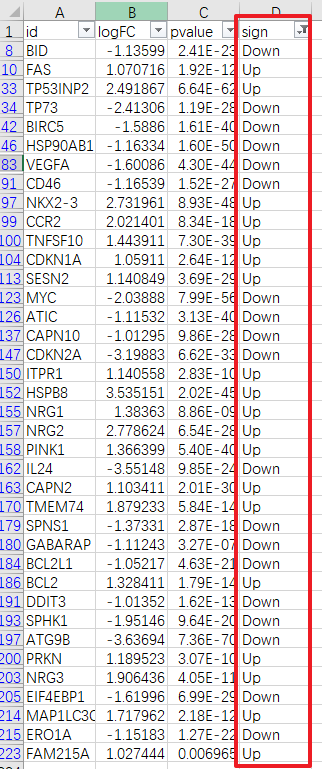
第3步,参考Figure 1b绘制的第4步,在第一行加入样本信息,把差异表达ARGs表达谱数据整理成仙桃简易数值热图要求格式: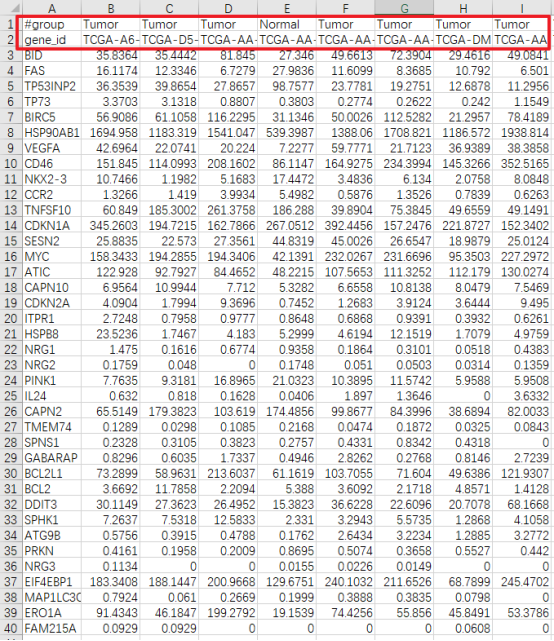
点击工具选择栏【分析工具】→点击左侧导航栏【表达差异】→点击右侧导航栏简易数值热图→上传第5步整理好的数据→点击【验证】→设置【聚类(顺序)/分割】→修改上注释颜色→点击【确认】→下载热图图片。
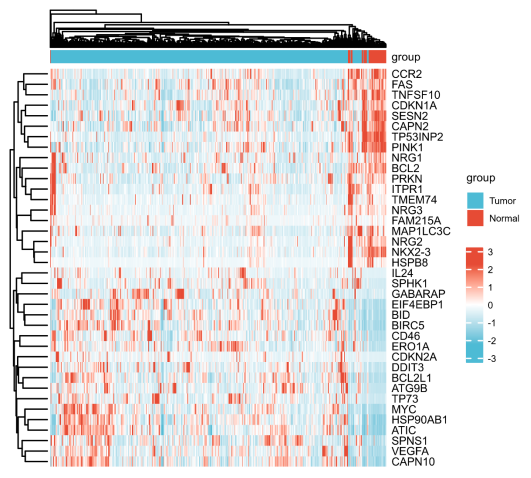
Figure 1c| ARGs表达分组比较图(Tumor vs Normal))点击工具选择栏【分析工具】→点击左侧导航栏【表达差异】→点击右侧【 [云]疾病vs非疾病】→选择云端数据集→输入差异表达ARGs分子→设置【误差线类型】→设置【X轴旋标注转45度】→设置图片【宽度】→点击【确认】→下载分组比较图。


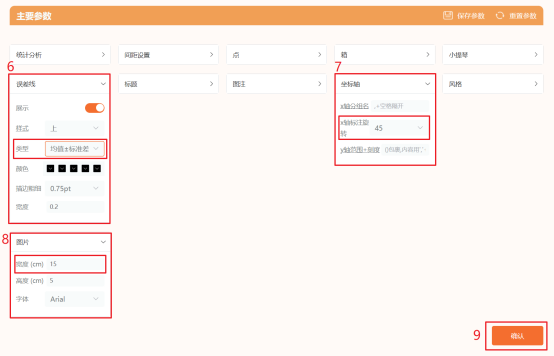
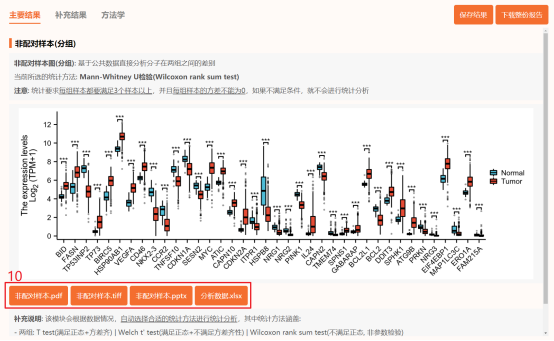
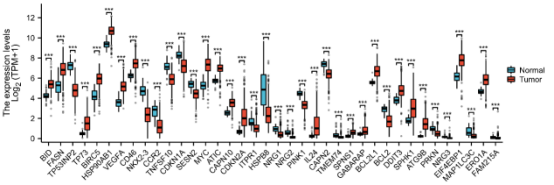
Figure 2| 差异表达ARGs的GO/KEGG富集分析Figure 2b| GO联合logFC富集分析-气泡图Figure 2c| KEGG联合logFC富集分析-圈图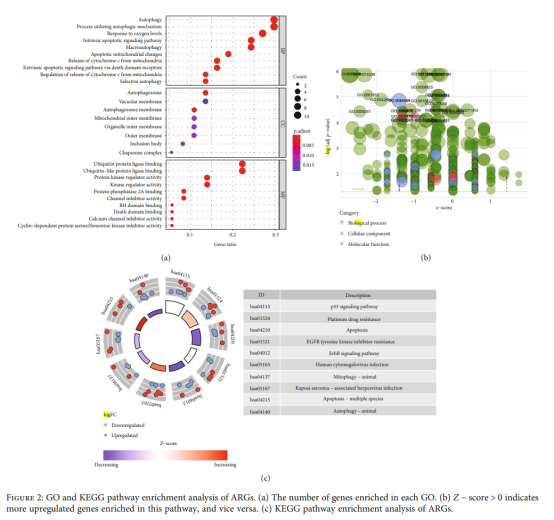
根据火山图绘制得到的差异表格整理差异表达ARGs列表,如下所示:
点击仙桃生信工具上方的工具选择栏【分析工具】→点击左侧导航栏【功能富集】→点击右侧导航栏【GOKEGG】中的【[GOKEGG]分析】→上传第1步的数据→点击【验证】→【富集参数】选择【全部GO】→默认其他参数→点击【确认】→保存结果。
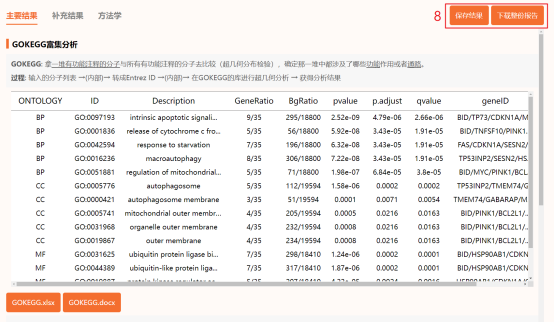
点击仙桃生信工具上方的工具选择栏【分析工具】→点击左侧导航栏【功能富集】→点击右侧导航栏【GOKEGG】中的【[GOKEGG]气泡图】→选择第2步保存的数据→默认主要参数→点击【确认】→下载图片。

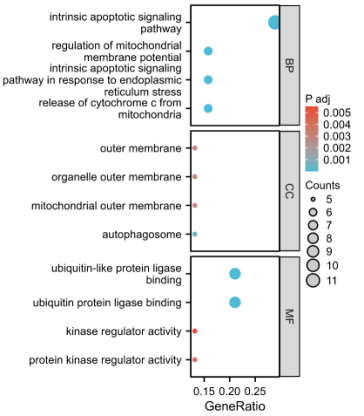
Figure 2b| GO联合logFC富集分析-气泡图
根据火山图绘制得到的差异表格整理差异表达ARGs的id和logFC数据,如下所示: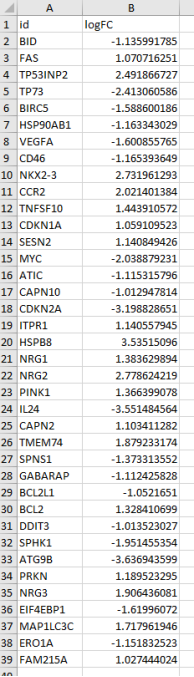
点击仙桃生信工具上方的工具选择栏【分析工具】→点击左侧导航栏【功能富集】→点击右侧导航栏【GOKEGG】中的【[GOKEGG联合logFC]分析】→选择第1步保存的数据→点击【验证】→【富集参数】选择【全部GO】→默认其他参数→点击【确认】→保存结果。

点击仙桃生信工具上方的工具选择栏【分析工具】→点击左侧导航栏【功能富集】→点击右侧导航栏【GOKEGG】中的【[GOKEG联合logFC]气泡图】→选择第2步保存的数据→默认主要参数→点击【确认】→保存结果。
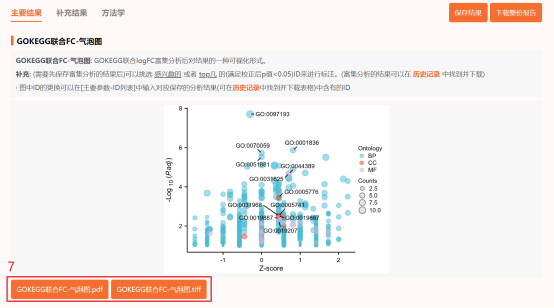
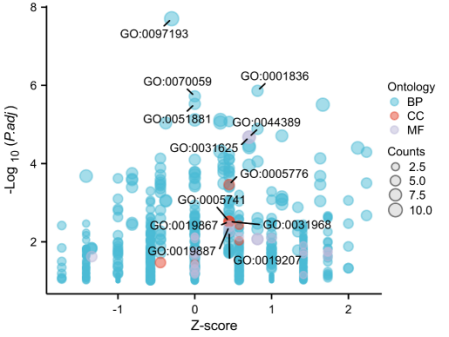
Figure 2c| KEGG联合logFC富集分析-圈图)复现:第1步,参考Figure 2b的第1步和第2步,在GOKEGG联合FC分析中,【富集参数】选择【KEGG】。第2步:点击仙桃学术工具上方的工具选择栏【分析工具】→点击左侧导航栏【功能富集】→点击右侧导航栏【GOKEGG】中的【[GOKEG联合logFC]圈图】→选择第1步保存的数据→默认主要参数→点击【确认】→保存结果。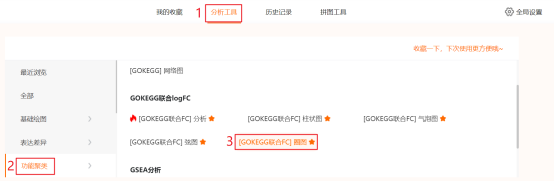

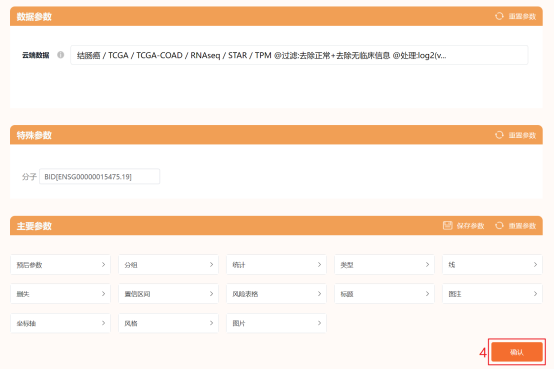
第1步:通过仙桃学术下载TAGA-COAD临床数据点击工具选择栏【分析工具】→点击左侧导航栏【临床意义】→点击右侧预后类带[云]标识的基于云端数据的分析模块,这里以【[云] 生存曲线(KM图)】为例→默认所有参数→点击【确认】→点击【方法学】→点击【百度云超链接】并复制提取码→在新窗口中粘贴提取码并点击【提取文件】→选择文件并点击【下载】。
提取下载的临床数据中的COAD患者的【sample_id】,【patient.vital_status】,【patient.days_to_death】和【patient.days_to_last_followup】四列数据并根据【patient.days_to_death】和【patient.days to_last_followup】整理出【OS time】(如果patient.vital_status是Alive,OS time提取【patient.days to_last_followup】列数据,如果patient.vital_status是Dead,提取【patient.days_to_death】列数据)→根据【patient.vital_status】整理出event数据(Alive定义未0,Dead定义为1)→加上差异表达ARGs的表达谱,数据就整理好了,如下所示: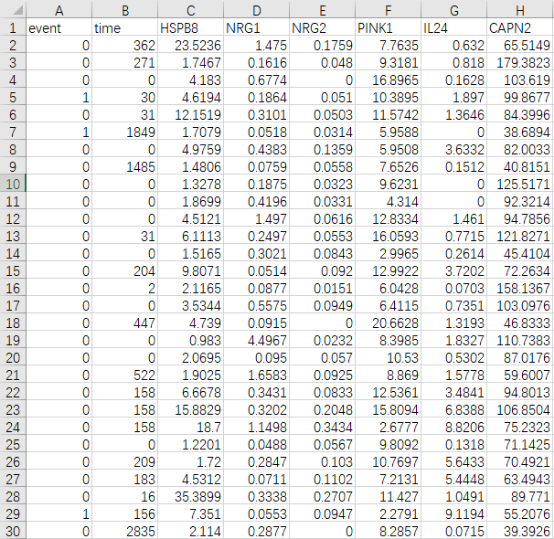
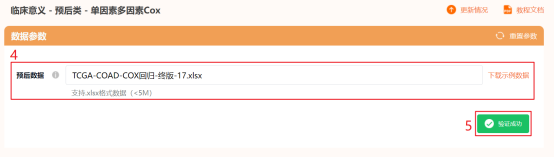
PS:根据仙桃单因素多因素Cox的数据要求,再查看一下有无时间为负值的情况,要求time以天计算,且不能出现负值和非数值的情况;目前,仙桃只能限制22列,我们一共有38个AGRs作为变量,所以需分两次进行,后期仙桃会更新这部分功能。点击仙桃学术工具上方的工具选择栏【分析工具】→点击左侧导航栏【临床意义】→点击右侧导航栏【预后类】中的【单因素多因素Cox】→上传第2步整理好的数据→默认主要参数→点击【确认】→保存结果,下载【Cox回归结果.xlsx】和【Riskscore.xlsx】。

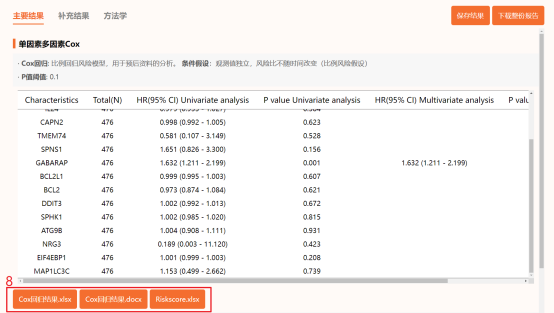
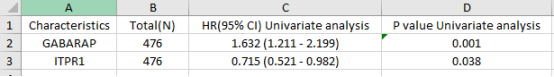
因为筛出的对OS总生存期有显著影响的分子数量太少,所以提示错误,这里没能复现出森林图。我们可以放上Cox回归分析三线表。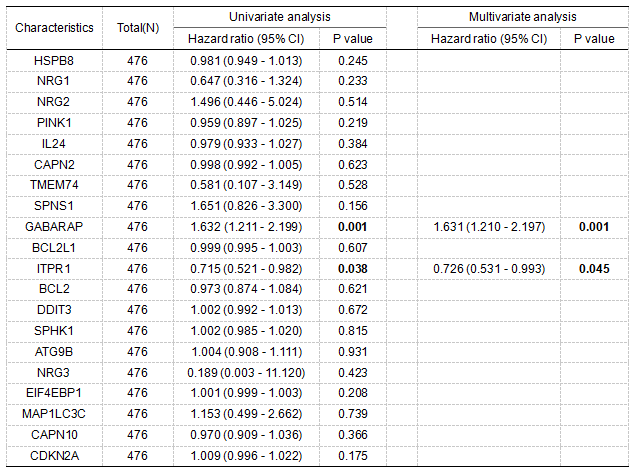
Figure 4a| ARGs在高/低风险组中的表达热图
Figure 4b| ARGs在高/低风险组中的风险得分
Figure 4c| ARGs在高/低风险组中的生存时间
Figure 4d| ARGs表达分组比较图(高/低风险组))
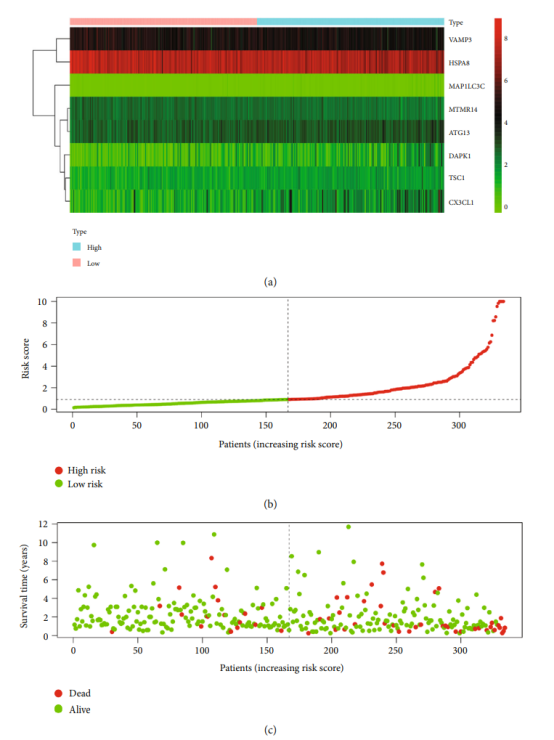
风险得分、生存结局、风险分组和热图,包含了Figure的a-c。根据Cox回归得到的RiskScore数据进行整理,把最后的RiskScore列移到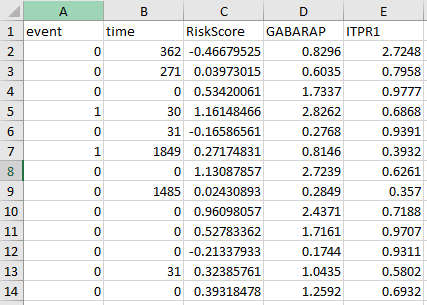
点击仙桃学术工具上方的工具选择栏【分析工具】→点击左侧导航栏【临床意义】→点击右侧导航栏【预后类】中的【风险因子图】→上传第1步整理好的→默认主要参数→点击【确认】→下载图片。
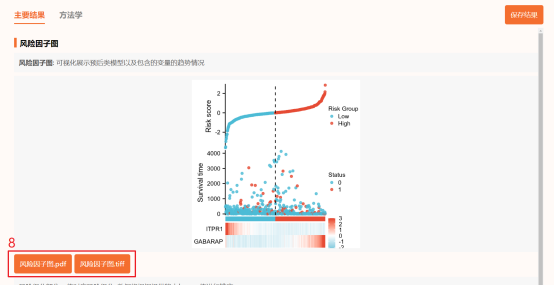
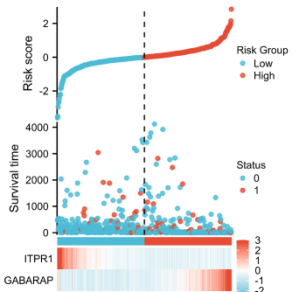
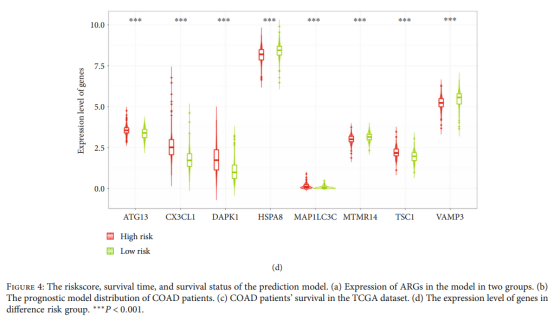
Figure 4d| ARGs表达分组比较图(高/低风险组)作者在这里用分组比较图展示了预后模型筛选出的基因在COAD和正常样本中的表达差异。我们选取COX回归模型中p值较低的两个基因给大家演示一下:第1步:根据Riskscore的中位数将样本分为高/低风险组,整理数据如下所示: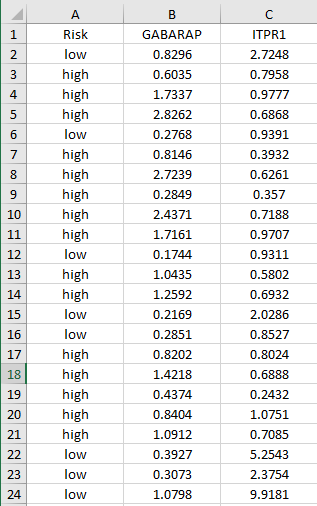
点击仙桃学术工具上方的工具选择栏【分析工具】→点击左侧导航栏【基础绘图】→点击右侧导航栏【类别比较】中的【分组比较图】→上传第1步整理好的→选择只展示【箱式图】→设置【误差线类型】→添加【y轴标题】→默认其他主要参数→点击【确认】→下载图片。
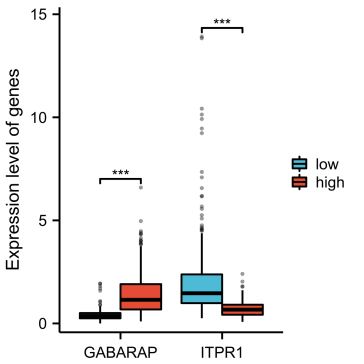
Figure 5a| 预后模型-列线图(基于Cox单因素回归结果)Figure 5b| 预后模型-列线图(基于Cox多因素回归结果)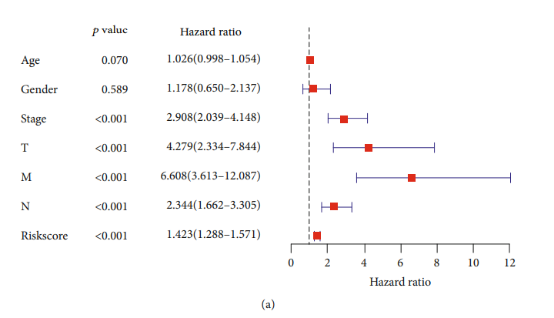
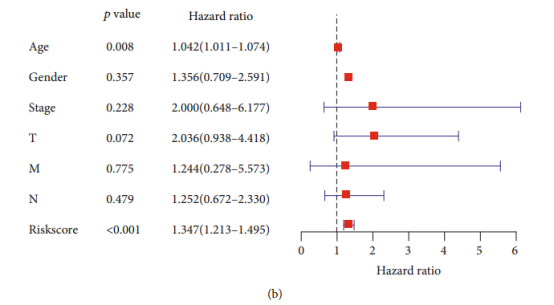
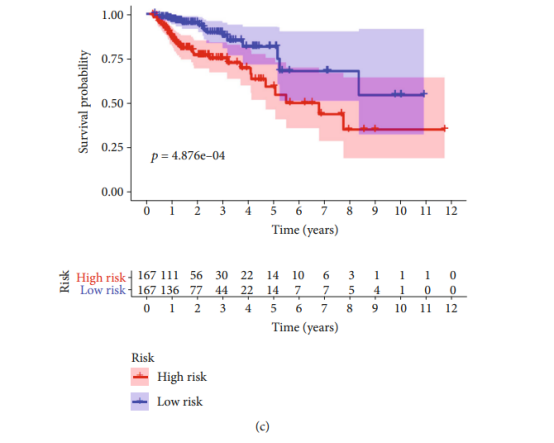
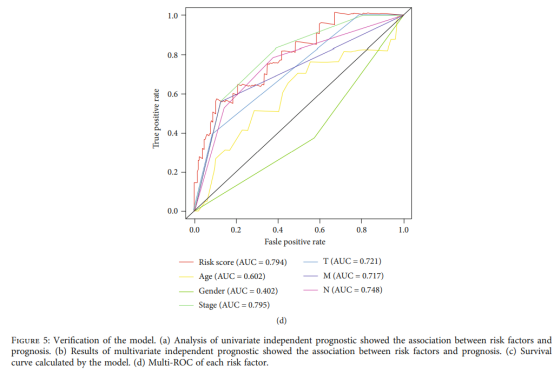
Figure 5是对模型的验证,原文用了结合了列线图、临床数据和风险因子的森林图、K-M生存曲线以及ROC曲线。
一般来说ROC曲线用于评价模型的区分能力,而只评估预后模型区分能力是不够的,还需要评估其准确度,而预后校准曲线就用来评估预后模型的准确度,也可以说校准度的重要方式。接下来我们就来完成时间依赖ROC曲线和预后校准曲线。根据Cox回归后的数据进行整理,去掉RiskScore列,如下所示: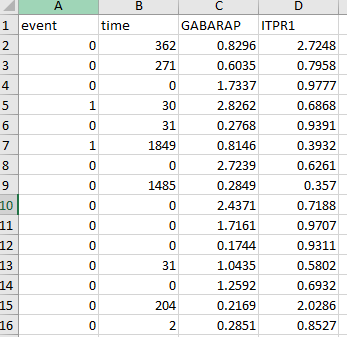
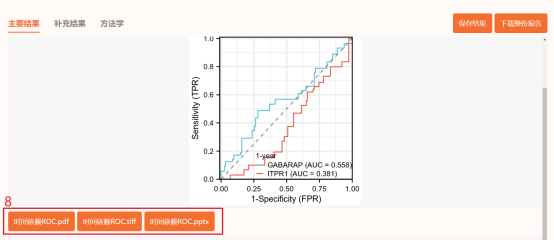
点击仙桃学术工具上方的工具选择栏【分析工具】→点击左侧导航栏【临床意义】→点击右侧导航栏【预后类】中的【时间依赖ROC】→上传第1步整理好的数据→点击【验证】→默认主要参数→点击【确认】→下载时间依赖ROC图片。
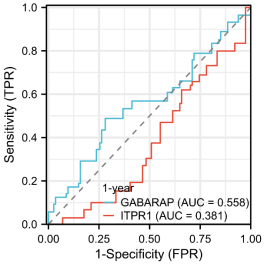
根据Cox回归后的数据进行整理,去掉RiskScore列,如下所示:
点击仙桃学术工具上方的工具选择栏【分析工具】→点击左侧导航栏【临床意义】→点击右侧导航栏【预后类】中的【预后校准曲线】→上传第1步整理好的→默认主要参数→点击【确认】→出现成功提示,点击【确定】→在【历史记录】中查看和下载预后校准曲线。

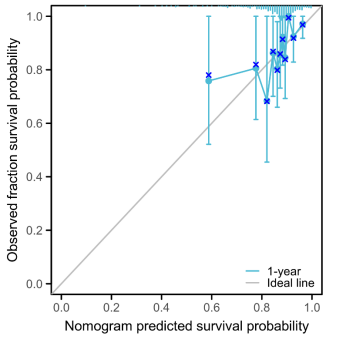
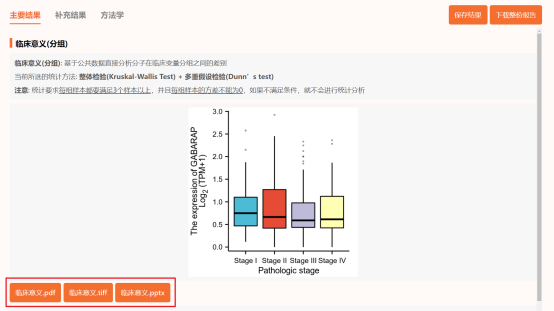
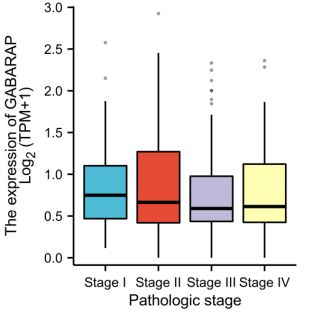
Figure 6| 临床特征分组比较图(ARGs预后模型筛选出的显著影响预后的因子和临床特征的相关性)。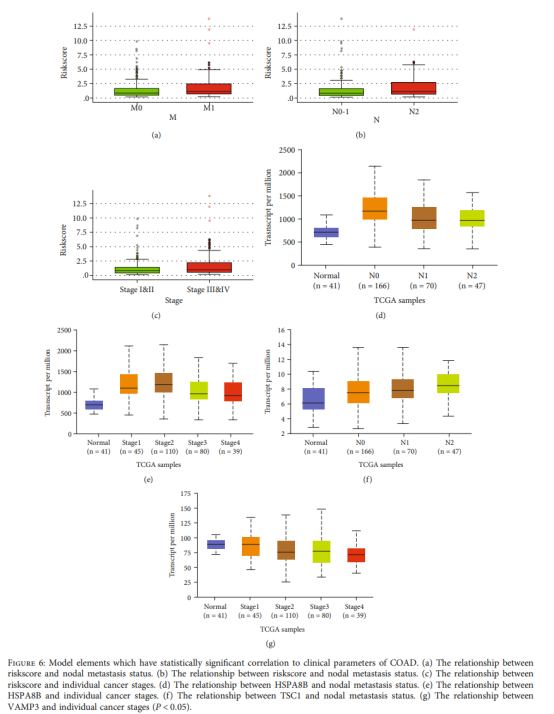
点击仙桃学术工具上方的工具选择栏【分析工具】→点击左侧导航栏【临床意义】→点击右侧导航栏【临床相关】中的【[云]临床意义(分组)】→选择数据集→输入分子GABARAP→选择【临床变量】→设置分组→默认主要参数→点击【确认】→下载临床意义(分组)图。
好啦,以上就是本篇文章复现的所有内容,这里最后再给大家小结一下,文中整体思路万变不离其宗, 从最开始的表达差异,接下来进行GO和KEGG富集分析,最后构建了预后模型并对其进行验证,并用模型筛选出的因素比较该因素在不同临床特征间的差异。最后说一下复现结果和原文结果一致性的问题,比如复现的结果与原文不一致。其实使用R语言分析,数据的清洗过程、使用的R包算法、阈值、剔除样本的标准等等,任何一个参数不一致都可能导致结果的不同。我们不可能保证和原文使用的R包算法、阈值、剔除样本完全一样。在复现过程中,这些参数甚至可能和原文差异较大,这也会导致复现结果和原文不同。这里还有几个坑,第一:HADb数据库下载得到的ARGs并不是232个,而是222个,因为有10个基因是重复的。第二:HADb数据库下载得到的ARGs有的Gene Symbol在表达谱中找不到,但实际上在Gene Card中查询,会发现其他的名字,再到表达谱中查询就可以找到了。因为基因名不是一成不变的,随着我们对基因更加深入地了解,基因名也会更新。由此可见,发表的文章中也会出现错误,也可能注意不到一些很细节的部分。所以大家在复现的时候,不要刻意追求和原文结果的完全一致,这个是很难的。R包算法、阈值、剔除样本的原因,也有文章可能出现的小错误。也有小伙伴可能担心:如果和原文结果不一致,我怎么知道是不是自己做错了?仙桃工具的靠谱也在于我们已经将数据清洗、质控在后台都做好了,所以大家使用云端数据是完全没有问题的,有了仙桃工具,大家主要是学会如何解读结果以及串成一个完整的故事,我们的初衷也是希望大家能够避开R语言就可以进行生信分析。祝愿大家都能够早日发表自己的生信文章,更多教学内容大家可以参照往期复现推文,也可以加入我们的仙桃陪伴营,包括但不限于生信入门方法论、生信文献泛读与精读、生信常见图表解读与实操、生信课题设计方法、文章复现课(直播和文字版)、写作课等,感兴趣的小伙伴千万不要错过啦~也可以扫下方二维码,加入导学营免费体验哦~(已加的同学不用重复加入)